
Omluva č. 1: Blog měl v poslední době extrémně dlouhé prodlevy mezi články. Doufám, že se mi s tím povede něco udělat.
Omluva č. 2: Několik posledních článků (dinosauři v pražské zoo, retropozony a fylogeneze) si pro sebe zabrali žijící ptáci (či jejich prsty) a poměrně technické články o embryologii či molekulární fylogenetice. Tento článek tuto sérii uzavírá, pak přijdou na řadu opět fosilie.
1. Úvod
O příbuzenských vztazích pěvců k ostatním ptákům jsem psal poměrně nedávno, a to v souvislosti se studií, která se jim snažila přijít na kloub pomocí vzácných genomových změn (Suh et al. 2011). Samozřejmě ale zdaleka nešlo o jediný pokus svého druhu – ve skutečnosti je toto téma v rámci ptačí fylogenetiky jedním z nejvděčnějších. Není divu. Pěvci (Passeriformes) jsou silně odvozená skupina, charakteristická mj. velkými mozky a metabolizmem mimořádně rychlým dokonce i na ptačí poměry. Krom toho se v nich skrývá velká část ptačí diverzity – zahrnují něco přes polovinu žijících ptačích "druhů", byť sčítat taxonomické kategorie (ať už jakékoli) je poměrně neuspokojivý způsob, jak diverzitu kvantifikovat. Mezi pěvce rovněž patří jeden z nejdůležitějších modelových organizmů z řad ptáků, zebřička pestrá (Taeniopygia guttata), která např. pro takovou neurobiologii představuje totéž, co pro embryologii kuře.
V minulém článku jsem stručně zrekapituloval, co o pozici pěvců říkají genetická data, a zmínil jsem i skutečnost, že s nástupem fylogenomických studií (analyzujících data z více genů současně) vyvstala jakási opatrná shoda. S tou se ale odmítají spokojit Wang et al. (2011), kteří ve své nové studii, nacházející se zatím v "in press" fázi žurnálu Molecular Biology and Evolution, poukazují na fakt, že ani vztahy podpořené velice rozsáhlými analýzami nejsou zdaleka tak pevné, jak by se mohlo zdát. Příkladem mohou být třeba žebernatky (Ctenophora), na jejichž postavení se neshodnou ani rozsáhlé molekulární datasety. Obecně podobné potíže nastávají v situaci, kdy je taxon, o jehož pozici se zajímáme, součástí nějaké velké vývojové radiace, při níž se vývojové větve štěpily tak rychle, že se na nich nestačily zafixovat žádné významné molekulární a morfologické znaky, podle nichž by dnešní fylogenetici mohli pořadí odštěpování zrekonstruovat. Špatnou zprávou je, že tento popis dokonale sedí na skupinu Neoaves, zahrnující většinu žijících ptáků – včetně pěvců. Wang et al. (2011) tedy navrhují vzít v úvahu všechny dosud navržené hypotézy a otestovat je rozborem nezávislých, dosud nevyzkoušených dat.
Jaká data by to měla být? Zdá se, že morfologie nám v případě Neoaves moc nepomůže. V předešlých 200 letech k tomu konec konců měla příležitostí víc než dost, a i Livezey & Zusi (2007), kteří přišli s bezprecedentním pokusem analyzovat fylogenetickými metodami téměř 3000 morfologických znaků, stále vykryli některá zdiskreditovaná uskupení, jako např. "klad" složený z potápek a potáplic nebo monofyletické "krátkokřídlé" sensu lato (tj. včetně perepelů – ve skutečnosti odvozených racků – a mesitů, kteří zase zřejmě mají blíž k potápkám a plameňákům). Pak se nabízí možnost použít sekvenční data. Jenže odkud? Stromy odvozené z jednotlivých genů (viz např. četná srovnání v Cracraft et al. 2004) jsou od sebe tak odlišné, že Poe & Chubb (2004) nebyli schopni ani vyvrátit předpoklad, že se každý gen vyvíjel zcela nezávisle na ostatních (což je vedlo k dosti pesimistické hypotéze rozebrané podrobněji v předchozím článku, podle níž nepůjde bazální příbuzenské vztahy Neoaves rozřešit se sebevětším množstvím dat). Chojnowski et al. (2008) ukázali, že to není pravda a že stromky založené na různých lokusech vykazují větší než náhodnou shodu; zároveň také navrhli, že do budoucna bude lepší zaměřit se na rychleji se vyvíjející jaderné introny (tedy nekódující sekvence genů, které se vystřihují z mRNA) a osekvenovat jich tolik, kolik jen bude možné. 16 kilobází je minimum; ideálně by to pak mělo být více než 32 – právě taková přitom byla velikost datové matrice v dosud největší sekvenční fylogenetické analýze ptáků (Hackett et al. 2008).
2. Introny, jejich potenciál, a jak je analyzovat
Wang et al. (2011) se tedy rozhodli, že nejvhodnějším zdrojem dat k rozřešení fylogenetického postavení pěvců budou jaderné introny. Jenže jejich použití s sebou také nese hned několik problémů: především na tento zdroj dat spoléhaly už předchozí analýzy (Hackett et al. 2008; v menší míře Ericson et al. 2006), jejichž výsledky se nyní mají testovat. Této cirkulárnosti je třeba se nějak vyhnout, ale jedno jednoduché řešení je naštěstí nasnadě: vytvořit zcela nový, nezávislý dataset z dosud neanalyzovaných intronů a pomocí něj tak zatím navržené hypotézy přezkoušet. Další problémy jsou metodologické. Introny se obecně vyvíjejí jednodušším způsobem než exony a nepřekládané oblasti (UTRs, myšlen 5'-konec ["čepička"] a 3'-konec ["ocásek"] molekuly mRNA) a vykazují např. menší rozdíly v substituční rychlosti mezi jednotlivými páry bází kodonu (Bonilla et al. 2011).* Jenže je s nimi také celá řada problémů: např. s "alignmentem", tedy přiřazováním odpovídajících si pozic v sekvencích od různých taxonů. Dalším problémem je substituční saturace, tedy zjednodušeně řečeno ztráta fylogenetické informace způsobená příliš rychlými mutacemi.
Aby se s těmito problémy vypořádali, vyzkoušeli Wang et al. (2011) několik strategií. Nejdůležitější je hlavně správně přiřadit homologické sekvence, tedy vědět, co se má s čím srovnávat. Autoři se o to pokusili jednak manuálně, jednak k tomu využili specializovaný software. Důkazem, že Wang a spol. mysleli skutečně na vše, je i jejich pojistka proti vlastním biasům, takže před manuálním alignmentem náhodně proházeli taxony a jejich jména skryli za náhodně vygenerované kódy, které nerozlouskli sami, ale přenechali je vlastnoručně napsanému programu. Poté přišli s několika dalšími verzemi, z nichž některé sekvence vyloučili, hlavně pak rozsáhlé inzerce a delece, které nejsou fylogeneticky informativní, a místa, kde jednoznačné přiřazení není možné. Pak si napsali – resp. Edward L. Braun napsal – další program (v jakém zvráceném světě to žijeme: ornitologové programují a programátoři pitvají ptáky), který využili k vyloučení všech nukleotidů, které jsou přítomny jen u dvou či méně taxonů. Stejný postup uplatnili i Hackett et al. (2008). Wang a spol. rovněž zmiňují, že i když má manuální zarovnávání sekvencí své výhody (jelikož lidský mozek aspoň občas dokáže najít mezi dlouhými řadami Áček, Téček, Céček a Géček podobnosti lépe než jednoduchý algoritmus), otevírá prostor různým biasům, které není možné úplně potlačit ani výše zmíněnou rafinovanou metodou; takže krom vlastních mozků zaměstnali ptačími intronovými sekvencemi celkem 6 různých, k tomu určených programů, v nichž si ještě pohráli s různými nastaveními, aby zjistili jejich dopad na výsledky analýzy. Jakmile tedy byly všechny možné variace vhodného uspořádání sekvencí hotové, přišel čas je analyzovat. Ovšem místo toho, aby Wang et al. (2011) prostě jen našli stromek nejlépe odpovídající daným datům, si po analýze vynutili několik stromků s pěvci na určité konkrétní pozici, a teprve potom srovnali, nakolik tyto stromky odpovídají analyzovaným sekvencím. Nejen to, oni postoupili ještě dál a znovu provedli alignment, tentokrát tak, aby co nejlépe odpovídal zadaným stromkům.
Tím nejtechničtější část studie končí. Zbývá snad jen dodat, že autoři použili bayesovské metody a jejich dataset sestával z 30 lokusů (představujících nějakých 27 kilobází zarovnaných sekvencí) na 17 chromozomech, přičemž ani jeden z nich dosud v žádné fylogenetické studii nebyl použit. Tato data byla získána od 28 ptačích taxonů, záměrně totožných s těmi, které použili Hackett et al. (2008) – přibyl jen nestor kea (Nestor notabilis) a zebřička pestrá (Taeniopygia guttata). Díky tomu bylo v závěru studie možné oba datasety zkombinovat a vytvořit tak "megaset" čítající 49 lokusů a 59 kilobází. Přestože studie se zajímá o fylogenezi poměrně úzkého výseku Neoaves (blízkého příbuzenstva pěvců), hypotézy, které mají být testovány, se absolutně neshodnou, kde se tento výsek nachází. Vybrat outgroup z řad Neoaves tedy nebylo možné: taxony, které by přicházely v úvahu jako rozumní kandidáti v jedné hypotéze (třeba vodní ptáci nebo jeřábi podle Hackett et al. 2008) jsou součástí ingroup v hypotéze druhé (Sibley & Ahlquist 1990). Jako outgroup proto byli nakonec zvoleni dva zástupci prvního kladu, o němž je absolutně jisté, že stojí mimo skupinu tvořenou všemi zahrnutými taxony – čili drůbeže. Konkrétně šlo o čáju obojkovou (Chauna torquata) a kuře (Gallus gallus). Autoři také provedli spoustu testů, které dokazují, že jejich výsledky nijak nezkreslují dobře známé problémy s genetickými daty (base composition bias atd.).


3. Co testovat aneb kdo má k pěvcům nejblíž
Teď je nutné obrátit se k hypotézám, které mají být otestovány. V minulém článku jsem snad mohl vzbudit mylný dojem, že všechny hypotézy, které stojí za řeč, byly vyprodukovány v posledních 10 letech a opírají se o fylogeneticky analyzovaná sekvenční data. To ale samozřejmě není pravda. První hypotézy o postavení pěvců mezi žijícími ptáky lze vystopovat až ke slavné klasifikaci Huxleyho (1867), který pěvce – v jeho systému nazývané "Coracomorphae" – shrnul dohromady se skupinou tvořenou lelkovitými (Caprimulgidae), kolibříky (Trochilidae) a rorýsovitými (tehdy zvanými Cypselidae). Zajímavé je, že toto uskupení (nazvané Cypselomorphae) bylo na dlouhou dobu pozapomenuto a "lelci" byli spojováni spíš se sovami, ale dnes je zase zpátky a zdá se být jedním z nejlépe podložených příbuzenství v Neoaves.
Fürbringer (1888) prezentoval podstatně odlišnou hypotézu, a můžeme bez nadsázky říct, že od jeho dob už morfologie s ničím novým nepřišla. Fürbringer svou hypotézu pokrokově zachytil formou stromu (který jsem přetiskl na blogu), na němž měli pěvci nejblíž ke skupině složené z datlů (Picidae), medozvěstek (Indicatoridae), tukanů (Ramphastidae) a "vousákovitých" ("Capitonidae" – umělá skupina parafyletická vůči tukanům). Výslednou skupinu Fürbringer dokonce pojmenoval, a to jako Pico-Passeres. S Fürbringerem souhlasil i Lowe (1946), který dokonce navrhl včlenit čtyři výše uvedené skupiny do pěvců samotných jako "podřád" Pici. Stejnou fylogenetickou hypotézu podpořili i Mayr & Amadon (1951). Nejobvyklejší alternativou byli "srostloprstí" ("Coraciiformes"). Se vzájemným vztahem šplhavců (Piciformes: Pici + Galbulidae + Bucconidae) a "srostloprstých" to bylo vždycky poněkud komplikované: celá řada autorů pokládala leskovce (Galbulidae + Bucconidae) za blíže příbuzné "srostloprstým" (např. Olson 1985), a naopak dudkovité (Lowe 1946) či klad tvořený dudkovitými, dudkovcovitými a zoborožci (Olson 1985) za blízké příbuzné Pici. Dnes se zdá, že "srostloprstí" jsou skutečně parafyletičtí vůči monofyletickým šplhavcům (avšak opačně, než si myslel Olson; zoborožci a dudci k nim naopak mají dál než zbytek skupiny – viz Hackett et al. 2008), a že tedy dohady, zda mají k pěvcům blíž jedni nebo druzí, nemají žádnou cenu. Nicméně hypotézu o tom, že sesterským taxonem k pěvcům jsou pikokoraciani (zoborožci + datli a všechno mezi tím) jako celek, můžeme považovat za dobrý začátek.
Na konci 60. let a v letech 70. došlo v ptačí systematice ke dvěma významným změnám: nástupu explicitně fylogenetických metod a přesunu od morfologických znaků k molekulárním datům. Tyto dvě novinky se ale dlouho vyvíjely mimoběžně, takže využití parsimonie bylo omezeno na morfologii a molekulární data byla až do 90. let analyzována často sice numerickými, ale relativně primitivními, na distancích založenými metodami typu UPGMA. Snahy analyzovat genetická data distančními metodami vyvrcholily slavnou prací Sibleyho a Ahlquista (1990), která nabízí další hypotézu k otestování. Výše uvedený obrázek ji zachycuje dosti nepřesně, protože zobrazuje jen ty taxony, jejichž pozici testovali také Wang et al. (2011). Sibleyho a Ahlquistovu bichli sice nemám, ale stromek z ní jsem viděl přetištěný v několika jiných publikacích (Cracraft et al. 2004; Livezey & Zusi 2007) a na něm je sesterskou skupinou pěvců klad složený ze většiny tradičních "krátkokřídlých", dále z holubů, dlouhokřídlých, stepokurů, denních dravců ("Falconiformes"), potápek, faetonů, různě rozptýlených "brodivých" a "veslonohých", trubkonosých, tučňáků a potáplic.
Hypotézy, které vyvstaly, když se ke konci 90. let situace přece jen změnila k lepšímu a i molekulární data se začala analyzovat fylogeneticky, jsem už popsal v minulém článku. Zopakuji tedy jen, že dva největší takové rozbory (Ericson et al. 2006; Hackett et al. 2008) shodně odhalily jako nejbližší žijící pěvců papoušky a jako druhé nejbližší příbuzné sokolovité.
Výčet zbývá uzavřít s asi posledním z velkých zdrojů dat, totiž s mitochondriální DNA. Ta v 90. letech podporovala dosti neuvěřitelnou hypotézu, podle níž byli právě pěvci – a nikoli paleognáti – sesterskou skupinou ke všem zbývajícím ptákům (Mindell et al. 1997; Härlid & Arnason 1999), což se ukázalo být jen artefaktem přitahování dlouhých větví (viz níže). Od té doby přišla řada důvěryhodnějších mtDNA analýz. Brown et al. (2008) použili neúplné mitochondriální sekvence (4594 párů bází) od úctyhodného množství taxonů (135) a v jejich výsledcích byli pěvci stále pěkně bazální: vyšli jako sesterská skupina všech ostatních neoavianů; bazálnější tedy byla už jen drůbež a paleognáti. To Pratt et al. (2009) použili kompletní mitochondriální genomy (přes 13 kilobází), ale od omezeného počtu taxonů (45). Vyšel jim pro změnu sesterský vztah s kukačkami, který už dříve analýzou mtDNA odkryli Sorenson et al. (2003) a kombinovaným rozborem morfologických znaků a tří jaderných genů Mayr et al. (2003).
4. Psittacopasserae znovu na scéně
A výsledky? Nic překvapivého. Byly potvrzeny výsledky Hackett et al. (2008), a to nejen v oblasti blízkých pěvčích příbuzných, ale i v topologii "pozemních ptáků" (landbirds – slib napsat o nich něco víc stále trvá...) obecně; jen pozici denních dravců (Accipitriformes) a sov (Strigiformes) nešlo ověřit. Tyto výsledky silně odporují Sibleymu a Ahlquistovi (1990) i topologiím založeným na mtDNA. Livezey & Zusi (2007) se dostali blíž a jejich strom jakousi verzi pozemních ptáků snad i ukazuje; zatímco podle nich ale mezi potomky posledního společného předka sokolovitých a šplhavců patří i Huxleyho cypselomorfové a kukačky, podle Hackett et al. (2008) ani Wang et al. (2011) tomu tak není. Ti druzí dokonce odkryli pozemní ptáky se 100% posteriorní pravděpodobností. Stejná je i pravděpodobnost existence kladu zahrnujícího trogony, zoborožce a dudky (Bucerotes), "jádro srostloprstých" a šplhavce, který rovněž odporuje Livezeyově a Zusiho hypotéze. V ní takový klad zahrnuje ještě pěvce a myšáky.
Wang et al. (2011) dále potvrdili existenci kladu (seriemy + sokolovití + papoušci + pěvci), který spolu s Boydem (2011) nazývám Passerimorphae, a existenci kladu (papoušci + pěvci), který Suh et al. (2011) nedávno nazvali Psittacopasserae. Ten ovšem nemá nijak vysokou podporu: bootstrapovou jen okolo 30% a posteriorní pravděpodobnost (která je obvykle vyšší než bootstrap, takže podporu trochu "nafukuje") 70%. U podobného typu analýz, který využívá zřetězené sekvence mnoha různých lokusů, ovšem můžeme pravděpodobnost nějaké konkrétní topologie určit i jiným způsobem, a to tak, že se podíváme na to, které geny ji odkrývají a které jí naopak odporují. Tento přístup hojně využíval při svém subjektivním hodnocení dosud provedených analýz např. Mayr (2011). Jeho aplikace na nynější studii ukáže, že z 30 lokusů jich celých 28 vykrylo klady Passerimorphae a Psittacopasserae buď v nejvěrohodnějším stromě (= stromě, který je optimální podle metody maximum likelihood), v bootstrapovém konsenzuálním stromě**, nebo v obou z nich.
Rozdíl oproti dřívějším analýzám (Hackett et al. 2008; Suh et al. 2011) spočívá hlavně v tom, že sokolovití zde mají blíž seriemám než papouškům a pěvcům; jinak řečeno, nebyl nalezen klad Eufalconimorphae. V tom se naopak – jak podotkl Alexander Suh na DML – shodují s jednou z dílčích analýz Ericsona et al. (2006), v níž autoři strom zakořenili s drůbeží (Galloanserae) namísto paleognátů. Takový výsledek je sice intuitivní (seriema a sokol jsou si asi podobní více, než sokol a kakadu), ale také krajně nedůvěryhodný. Suh et al. (2011) podpořili Eufalconimorphae 7 retropozonovými inzercemi, největším počtem, jaký v jejich analýze nějaký klad obdržel (jinak řečeno, z hlediska retropozonů máme u monofylii eufalkonimorfů víc důkazů, než např. o monofylii pěvců). Po zahrnutí dat z Hackett et al. (2008) se ovšem Eufalconimorphae znovu objevilo; současně také prudce vzrostla podpora pro Psittacopasserae (na téměř 80%).
Zajímavé také je, že rozdíly v zarovnání sekvencí měly jen velmi malý dopad na výsledky analýzy. Ať už bylo ponecháno původní manuální zarovnání nebo z něj byly odstraněny výše zmíněné problematické úseky, výsledky si stále byly hodně podobné: odstraněné části poskytovaly stejný signál jako ty, které byly ponechány. Stejně tak automaticky vygenerovaná zarovnání stále vyhazovala podobnou topologii, a to s jedinou výjimkou u programu ClustalW2, který je ovšem "known to perform poorly" (Wang et al. 2011:18). Autoři argumentují, že jejich výsledky jednoznačně dokazují, že Chojnowski et al. (2008) měli pravdu, když označili introny za ideální zdroj dat pro fylogenetické analýzy založené na sekvencích, a že výsledky dosud provedených analýz založené na intronech (hlavně pak Hackett et al. 2008) nebyly zkreslené problémy se zarovnáním. Stále ovšem zbývá řada jevů, které mohly výsledky zkreslit, a Wang et al. (2011) se na ně připravili.
Jedním z nich je nechvalně známé přitahování dlouhých větví (Long Branch Attraction, LBA). Taxony, které se z molekulárního hlediska vyvíjejí rychleji než jiné (= v jejichž genomu častěji dochází k bodovým mutacím), budou na dané pozici sdílet tentýž nukleotid s větší pravděpodobností, než taxony s pomalou molekulární evolucí, a to bez ohledu na to, jaké je jich skutečné příbuzenství. Špatnou zprávou je, že zrovna pěvci dlouhou větev skutečně mají, a jak jsem napsal výše, některé fylogenetické rozbory to dokázalo pěkně zmást. Rozbor Wanga et al. (2011) to ale zřejmě příliš neovlivnilo, jelikož pěvci se zde shlukují s taxony, jejichž rychlost molekulární evoluce je pod mediánem: s papoušky, sokolovitými a seriemami.
Jelikož si ale autoři chtějí být jistí opravdu vším, prověřili ještě, zda náhodou přitahování dlouhých větví nemůže aspoň za monofylii Psittacopasserae, protože větev papoušků je pořád delší než větev sokolů a seriem. Metod, jak se s LBA vyrovnat, je několik. Může se použít specifický evoluční model***, který tyto rozdíly bere v úvahu. Wang et al. (2011) jeden takový použili a Passerimorphae i Psittacopasserae byly na výsledném stromě stále přítomny. Pokud je to možné, můžeme taky nahradit taxony s dlouhou větví těmi, jejichž molekulární evoluce je pomalejší. Zde to možné bylo: poměrně "pomalí" jsou bazální pěvec pokřovník (Acanthisitta) a bazální zpěvný lyrochvost (Menura), ale nic naplat – i když odstraníme z matice všechny ostatní, rychlejší pěvce, oba taxony se stále sdružují s papoušky a pak se sokolovitými a seriemami. Že by tato klady byla pouhými artefakty, nenaznačil ani test na saturaci: z té bylo v podezření 8 lokusů, jejichž odebrání opět výsledky nijak neovlivnilo. Závěr je jasný: data podporující blízkost papoušků a pěvců nesou pravý fylogenetický signál.

5. Myšáci, biogeografický původ pěvců a implikace pro Metaves
Další zajímavou částí studie je její zjevná neschopnost vypořádat se s myšáky (Coliidae). V článku o retropozonové studii Suha a spol. jsem napsal, že si tento klad "buduje reputaci fylogenetické hádanky srovnatelné i s proslulým hoacinem". Wang et al (2011) tento můj úsudek pouze potvrzují. Vzpomínáte na 2 lokusy, které nepodpořily sesterský vztah mezi pěvci a papoušky? Podle nich byly sesterskou skupinou papoušků myšáci, a teprve potom pěvci. Tento vztah podporuje také morfologie zadních končetin a gen ZENK (Chubb 2004), ostře mu ovšem odporují retropozony (Suh et al. 2011) a dosavadní sekvenční data (Ericson et al. 2006; Hackett et al. 2008). Ostatní geny v datasetu Wanga a spol. myšáky umisťují v sesterském vztahu k trogonům, sokolovitým, pěvcům (14% analýz), nebo psittakopasseranům či passerimorfům jako celku. Po vyloučení myšáků výrazně vzrostla stabilita všech kladů napříč stromem a zvýšila se i bootstrapová podpora passerimorfů a psittakopasseranů. Autoři ale nepovažují situaci z hlediska sekvenčních dat za zcela beznadějnou: problém s myšáky spočívá především v jejich dlouhé větvi, kterou by možná šlo zlomit důkladnějším taxon samplingem: pomoct by mohly třeba sovy (s nimiž se myšáci shlukli v Hackett et al. 2008), které do studie nebyly zahrnuty, jelikož pro příbuzenství pěvců nemají velký význam. Zatím ale nemůže být vyloučena možnost, že myšáci mají blízko k pěvcům – a asi i jakékoli jiné skupině pozemních ptáků.
Wang et al. (2011) se také zabývají biologickými důsledky sesterského vztahu mezi pěvci a papoušky, především vokálním učením. V této pasáži prakticky do detailů opakují to, co už před nimi na toto téma publikovali Suh et al. (2011): dosud se myslelo, že tuto schopnost nezávisle na sobě vyvinuly tři skupiny ptáků (papoušci, zpěvní pěvci a svišťouni), pod novou hypotézou se ale zdá logičtější, že první dvě ji zdědili od společného předka, což podporují i nedávná zjištění vokálního učení u některých křikavých. Zajímavější je to, co autoři o říkají o biogeografii. Sesterskou skupinou vůči zbytku pěvců (zahrnujícímu jak křikavé [Suboscines], tak zpěvné [Oscines]) jsou pokřovníkovití (Acanthisittidae), drobný, málo prozkoumaný klad špatných letců endemických pro Nový Zéland. Základní dělení papoušků je na klad zahrnující naprostou většinu diverzity celé skupiny, včetně kakaduovitých a arů, a na skupinu Strigopidae (česky zřejmě kakapovití), zahrnující největšího žijícího papouška – nelétavého, silně ohroženého kakapo sovího (Strigops) – a jednoho z nejinteligentnějších známých ptáků s extrémně rozvinutou schopností používat nástroje, nestora kea (Nestor). Zajímavé na nestorovi je ovšem také to, že stejně jako pokřovníkovití pochází z Nového Zélandu: že by právě zde žil společný předek celé skupiny Psittacopasserae? Podle Wanga a spol. je to pravděpodobné, vyloučit ale nelze ani to, že diverzifikace celé skupiny sledovala rozpad Gondwany, jak bylo navrženo např. pro "ratity" (Cracraft 2001; Johnston 2011). Molekulární hodiny se ale takovému scénáři zdají odporovat.
Poslední příležitost dozvědět se něco víc o příbuzenstvích pozemních ptáků představuje zkombinování původního datasetu se sekvencemi Hackettové a spol. ve stylu "víc genů víc ví". Hlavním cílem autorů bylo posoudit pomocí analýzy "independent evidence" dosavadní hypotézy; není ale bez zajímavosti z toho vzešlý strom srovnat s "total-evidence" stromem, který už nic netestuje, ale má k dispozici ještě nějaká data navíc. Výsledek ale není příliš překvapivý: topologie se přesně shoduje s tím, co odkryli Hackett et al. (2008) – s jedinou výjimkou, a tou je pozice lelků (Strisores). V kladogramu Hackettové a kolektivu jsou tito součástí Metaves, kontroverzního kladu, který navzdory tomu, že je založen doslova na pár desítkách nukleotidů v jediném intronu jediného jaderného genu, představuje zřejmě nejlepší hypotézu o bazálních divergencích Neoaves, jakou máme k dispozici. Odkryly ji totiž dvě dosud nejobsáhlejší molekulární analýzy (Ericson et al. 2006; Hackett et al. 2008) a žádná ji explicitně nevyvrátila, včetně té retropozonové (Suh et al. 2011). Jenže zatímco podle ní patří lelkové do Metaves a jeřábi, chřástalové či kukačky naopak spolu s pozemními ptáky do Coronaves, Wang a spol. odkryli klad složený z pozemních ptáků a lelků, který kukačky ani monofyletické jádro "krátkokřídlých" nezahrnuje.
Co tohle všechno znamená? Dost možná vůbec nic, jelikož analýze o podobné otázky vůbec nejde a spousta relevantních taxonů chybí, protože nám neříkají nic zajímavého o pozici pěvců (potáplice, dlouhokřídlí, dropové, holubi, stepokurové, svišťouni, mirandorniti a konec konců i spousta pozemních ptáků). Na druhou stranu ale zatím zdaleka nejobsáhlejší molekulární datová matrice vyprodukovala kladogram, který s hypotézou Metaves prostě nelze sloučit. Co se zbytku analýzy týče, klady Passerimorphae a (Picocoraciae + trogoni) mají ještě vyšší podporu než v Hackett et al. (83% a 79% vs. 64% a 71%), zatímco podpora Psittacopasserae zůstává stejná (78% vs. 77%). Důvodem jsou zřejmě opět zlotřilí myšáci. Ani kladogram vzešlý z kombinovaného datasetu nejeví známky přitahování dlouhých větví.

Celkově lze říct, že Wang et al. (2011) ukázali 1), které klady podpořené Hackettovou a kolektivem jsou nejpevnější a které si zaslouží další výzkum (byť bych si dovolil dodat, že vztahy mezi psittakopasserany, sokolovitými a seriemami asi nebudou tak problematické, jak autoři naznačují; 7 retropozonů je prostě hodně); 2) že studie a publikace, které zacházejí se stromkem z Hackett et al. pomalu jako se zjevenou pravdou (Hieronymus & Witmer 2010; Braun et al. 2011; Dyke & Kaiser 2011), což je přístup, který ornitologie zažila naposledy s tapisérií Sibleyho a Ahlquista, vzhledem k současnému stavu znalostí dobře dělají; 3) že Neoaves skutečně nejsou tvrdou polytomií, ale že i tuto mimořádně rychlou radiaci se povede s dostatečným množstvím molekulárních dat rozřešit. Takže nakonec docela optimistické zprávy.
6. Acknowledgments
Za zaslání fulltextu studie děkuji Alexanderu Suhovi a Janu Zrzavému.
*Každý kodon se skládá ze tří párů bází. V tom 3. dochází téměř vždy k substituci – změně "písmena" – častěji než v 1. a v 1. častěji než v 2. Pokud analýza tyto rozdíly nezapočítá, může podávat zkreslené výsledky: třetí kodon je např. vhodný k rekonstrukci mělkých vztahů (tedy těch, kde je společný předek analyzovaných taxonů poměrně nedávný), které nezasahují hluboko do minulosti; ale může podávat zavádějící výsledky u vztahů hlubokých.
**Neparametrický bootstrap je statistický test robustnosti výsledků analýzy. Jeho fylogenetická implementace v zásadě tahá z původního datasetu s opakováním, dokud nevytvoří nový dataset stejné velikosti ("pseudoreplikát"), v němž některé z původních znaků chybějí a jiné jsou zahrnuty vícekrát. Pro každý z množství pseudoreplikátů potom odhadne nový strom. Můžeme se buď dívat na to, v kolika takových stromech se vyskytuje uzel, jehož podporu testujeme, nebo z těchto stromků můžeme sestrojit strom konsenzuální, který může suplovat strom vytvořený na základě kompletní matrice. Velmi hezky popsáno zde.
***Evoluční model je (v tomto smyslu) popisem procesu nukleotidových substitucí – procesu nahrazování různých bází v DNA různými bázemi jinými. Každý aspekt substitučního procesu (např. rovnovážná četnost určité báze nebo relativní rychlost přechodu mezi danými dvěma bázemi) je v modelu reprezentován parametrem, jehož hodnotu analýza odhaduje spolu s tvarem stromu (topologií) a délkami větví (které udávají střední hodnotu počtu změn, k nimž na té které větvi došlo).
Zdroje:
Omluva č. 2: Několik posledních článků (dinosauři v pražské zoo, retropozony a fylogeneze) si pro sebe zabrali žijící ptáci (či jejich prsty) a poměrně technické články o embryologii či molekulární fylogenetice. Tento článek tuto sérii uzavírá, pak přijdou na řadu opět fosilie.
1. Úvod
O příbuzenských vztazích pěvců k ostatním ptákům jsem psal poměrně nedávno, a to v souvislosti se studií, která se jim snažila přijít na kloub pomocí vzácných genomových změn (Suh et al. 2011). Samozřejmě ale zdaleka nešlo o jediný pokus svého druhu – ve skutečnosti je toto téma v rámci ptačí fylogenetiky jedním z nejvděčnějších. Není divu. Pěvci (Passeriformes) jsou silně odvozená skupina, charakteristická mj. velkými mozky a metabolizmem mimořádně rychlým dokonce i na ptačí poměry. Krom toho se v nich skrývá velká část ptačí diverzity – zahrnují něco přes polovinu žijících ptačích "druhů", byť sčítat taxonomické kategorie (ať už jakékoli) je poměrně neuspokojivý způsob, jak diverzitu kvantifikovat. Mezi pěvce rovněž patří jeden z nejdůležitějších modelových organizmů z řad ptáků, zebřička pestrá (Taeniopygia guttata), která např. pro takovou neurobiologii představuje totéž, co pro embryologii kuře.
V minulém článku jsem stručně zrekapituloval, co o pozici pěvců říkají genetická data, a zmínil jsem i skutečnost, že s nástupem fylogenomických studií (analyzujících data z více genů současně) vyvstala jakási opatrná shoda. S tou se ale odmítají spokojit Wang et al. (2011), kteří ve své nové studii, nacházející se zatím v "in press" fázi žurnálu Molecular Biology and Evolution, poukazují na fakt, že ani vztahy podpořené velice rozsáhlými analýzami nejsou zdaleka tak pevné, jak by se mohlo zdát. Příkladem mohou být třeba žebernatky (Ctenophora), na jejichž postavení se neshodnou ani rozsáhlé molekulární datasety. Obecně podobné potíže nastávají v situaci, kdy je taxon, o jehož pozici se zajímáme, součástí nějaké velké vývojové radiace, při níž se vývojové větve štěpily tak rychle, že se na nich nestačily zafixovat žádné významné molekulární a morfologické znaky, podle nichž by dnešní fylogenetici mohli pořadí odštěpování zrekonstruovat. Špatnou zprávou je, že tento popis dokonale sedí na skupinu Neoaves, zahrnující většinu žijících ptáků – včetně pěvců. Wang et al. (2011) tedy navrhují vzít v úvahu všechny dosud navržené hypotézy a otestovat je rozborem nezávislých, dosud nevyzkoušených dat.
Jaká data by to měla být? Zdá se, že morfologie nám v případě Neoaves moc nepomůže. V předešlých 200 letech k tomu konec konců měla příležitostí víc než dost, a i Livezey & Zusi (2007), kteří přišli s bezprecedentním pokusem analyzovat fylogenetickými metodami téměř 3000 morfologických znaků, stále vykryli některá zdiskreditovaná uskupení, jako např. "klad" složený z potápek a potáplic nebo monofyletické "krátkokřídlé" sensu lato (tj. včetně perepelů – ve skutečnosti odvozených racků – a mesitů, kteří zase zřejmě mají blíž k potápkám a plameňákům). Pak se nabízí možnost použít sekvenční data. Jenže odkud? Stromy odvozené z jednotlivých genů (viz např. četná srovnání v Cracraft et al. 2004) jsou od sebe tak odlišné, že Poe & Chubb (2004) nebyli schopni ani vyvrátit předpoklad, že se každý gen vyvíjel zcela nezávisle na ostatních (což je vedlo k dosti pesimistické hypotéze rozebrané podrobněji v předchozím článku, podle níž nepůjde bazální příbuzenské vztahy Neoaves rozřešit se sebevětším množstvím dat). Chojnowski et al. (2008) ukázali, že to není pravda a že stromky založené na různých lokusech vykazují větší než náhodnou shodu; zároveň také navrhli, že do budoucna bude lepší zaměřit se na rychleji se vyvíjející jaderné introny (tedy nekódující sekvence genů, které se vystřihují z mRNA) a osekvenovat jich tolik, kolik jen bude možné. 16 kilobází je minimum; ideálně by to pak mělo být více než 32 – právě taková přitom byla velikost datové matrice v dosud největší sekvenční fylogenetické analýze ptáků (Hackett et al. 2008).
2. Introny, jejich potenciál, a jak je analyzovat
Wang et al. (2011) se tedy rozhodli, že nejvhodnějším zdrojem dat k rozřešení fylogenetického postavení pěvců budou jaderné introny. Jenže jejich použití s sebou také nese hned několik problémů: především na tento zdroj dat spoléhaly už předchozí analýzy (Hackett et al. 2008; v menší míře Ericson et al. 2006), jejichž výsledky se nyní mají testovat. Této cirkulárnosti je třeba se nějak vyhnout, ale jedno jednoduché řešení je naštěstí nasnadě: vytvořit zcela nový, nezávislý dataset z dosud neanalyzovaných intronů a pomocí něj tak zatím navržené hypotézy přezkoušet. Další problémy jsou metodologické. Introny se obecně vyvíjejí jednodušším způsobem než exony a nepřekládané oblasti (UTRs, myšlen 5'-konec ["čepička"] a 3'-konec ["ocásek"] molekuly mRNA) a vykazují např. menší rozdíly v substituční rychlosti mezi jednotlivými páry bází kodonu (Bonilla et al. 2011).* Jenže je s nimi také celá řada problémů: např. s "alignmentem", tedy přiřazováním odpovídajících si pozic v sekvencích od různých taxonů. Dalším problémem je substituční saturace, tedy zjednodušeně řečeno ztráta fylogenetické informace způsobená příliš rychlými mutacemi.
Aby se s těmito problémy vypořádali, vyzkoušeli Wang et al. (2011) několik strategií. Nejdůležitější je hlavně správně přiřadit homologické sekvence, tedy vědět, co se má s čím srovnávat. Autoři se o to pokusili jednak manuálně, jednak k tomu využili specializovaný software. Důkazem, že Wang a spol. mysleli skutečně na vše, je i jejich pojistka proti vlastním biasům, takže před manuálním alignmentem náhodně proházeli taxony a jejich jména skryli za náhodně vygenerované kódy, které nerozlouskli sami, ale přenechali je vlastnoručně napsanému programu. Poté přišli s několika dalšími verzemi, z nichž některé sekvence vyloučili, hlavně pak rozsáhlé inzerce a delece, které nejsou fylogeneticky informativní, a místa, kde jednoznačné přiřazení není možné. Pak si napsali – resp. Edward L. Braun napsal – další program (v jakém zvráceném světě to žijeme: ornitologové programují a programátoři pitvají ptáky), který využili k vyloučení všech nukleotidů, které jsou přítomny jen u dvou či méně taxonů. Stejný postup uplatnili i Hackett et al. (2008). Wang a spol. rovněž zmiňují, že i když má manuální zarovnávání sekvencí své výhody (jelikož lidský mozek aspoň občas dokáže najít mezi dlouhými řadami Áček, Téček, Céček a Géček podobnosti lépe než jednoduchý algoritmus), otevírá prostor různým biasům, které není možné úplně potlačit ani výše zmíněnou rafinovanou metodou; takže krom vlastních mozků zaměstnali ptačími intronovými sekvencemi celkem 6 různých, k tomu určených programů, v nichž si ještě pohráli s různými nastaveními, aby zjistili jejich dopad na výsledky analýzy. Jakmile tedy byly všechny možné variace vhodného uspořádání sekvencí hotové, přišel čas je analyzovat. Ovšem místo toho, aby Wang et al. (2011) prostě jen našli stromek nejlépe odpovídající daným datům, si po analýze vynutili několik stromků s pěvci na určité konkrétní pozici, a teprve potom srovnali, nakolik tyto stromky odpovídají analyzovaným sekvencím. Nejen to, oni postoupili ještě dál a znovu provedli alignment, tentokrát tak, aby co nejlépe odpovídal zadaným stromkům.
Tím nejtechničtější část studie končí. Zbývá snad jen dodat, že autoři použili bayesovské metody a jejich dataset sestával z 30 lokusů (představujících nějakých 27 kilobází zarovnaných sekvencí) na 17 chromozomech, přičemž ani jeden z nich dosud v žádné fylogenetické studii nebyl použit. Tato data byla získána od 28 ptačích taxonů, záměrně totožných s těmi, které použili Hackett et al. (2008) – přibyl jen nestor kea (Nestor notabilis) a zebřička pestrá (Taeniopygia guttata). Díky tomu bylo v závěru studie možné oba datasety zkombinovat a vytvořit tak "megaset" čítající 49 lokusů a 59 kilobází. Přestože studie se zajímá o fylogenezi poměrně úzkého výseku Neoaves (blízkého příbuzenstva pěvců), hypotézy, které mají být testovány, se absolutně neshodnou, kde se tento výsek nachází. Vybrat outgroup z řad Neoaves tedy nebylo možné: taxony, které by přicházely v úvahu jako rozumní kandidáti v jedné hypotéze (třeba vodní ptáci nebo jeřábi podle Hackett et al. 2008) jsou součástí ingroup v hypotéze druhé (Sibley & Ahlquist 1990). Jako outgroup proto byli nakonec zvoleni dva zástupci prvního kladu, o němž je absolutně jisté, že stojí mimo skupinu tvořenou všemi zahrnutými taxony – čili drůbeže. Konkrétně šlo o čáju obojkovou (Chauna torquata) a kuře (Gallus gallus). Autoři také provedli spoustu testů, které dokazují, že jejich výsledky nijak nezkreslují dobře známé problémy s genetickými daty (base composition bias atd.).

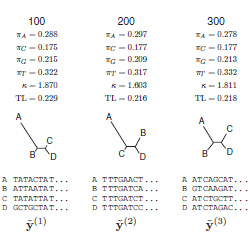
Tři možní kandidáti na nejbližší žijící příbuzné pěvců. Nahoře papoušci, zastoupení nestorem kea (Nestor notabilis); vlevo dole kukačky, na obrázku potom kukačka pokřovní (Cacomantis variolosus); vpravo dole Pici/Piciformes/Picocoraciae, reprezentovaní tukanem obrovským (Ramphastos toco). Hypotézy Sibleyho a Ahlquista i Browna a kolektivu jsem obrázky neilustroval: podle nich je totiž sesterskou skupinou pěvců valná většina nebo dokonce všichni Neoaves, takže by bylo poněkud těžké vybrat reprezentativního zástupce. (Modifikováno z upload.wikimedia.org)

Pět hlavních hypotéz o příbuzenských vztazích pěvců k ostatním ptákům, které byly získány DNA-DNA hybridizací (a) nebo fylogenetickou analýzou morfologických (b), jaderných (c) nebo mitochondriálních (d, e) dat. Zkratky: (CPBT) klad složený z trogonů, šplhavců a parafyletických "srostloprstých"; (PPFC) klad složený ze seriem, sokolovitých, pěvců a papoušků, odpovídající Passerimorphae sensu Boyd (2011). (Zdroj: Wang et al. 2011: Figure 1)
Teď je nutné obrátit se k hypotézám, které mají být otestovány. V minulém článku jsem snad mohl vzbudit mylný dojem, že všechny hypotézy, které stojí za řeč, byly vyprodukovány v posledních 10 letech a opírají se o fylogeneticky analyzovaná sekvenční data. To ale samozřejmě není pravda. První hypotézy o postavení pěvců mezi žijícími ptáky lze vystopovat až ke slavné klasifikaci Huxleyho (1867), který pěvce – v jeho systému nazývané "Coracomorphae" – shrnul dohromady se skupinou tvořenou lelkovitými (Caprimulgidae), kolibříky (Trochilidae) a rorýsovitými (tehdy zvanými Cypselidae). Zajímavé je, že toto uskupení (nazvané Cypselomorphae) bylo na dlouhou dobu pozapomenuto a "lelci" byli spojováni spíš se sovami, ale dnes je zase zpátky a zdá se být jedním z nejlépe podložených příbuzenství v Neoaves.
Fürbringer (1888) prezentoval podstatně odlišnou hypotézu, a můžeme bez nadsázky říct, že od jeho dob už morfologie s ničím novým nepřišla. Fürbringer svou hypotézu pokrokově zachytil formou stromu (který jsem přetiskl na blogu), na němž měli pěvci nejblíž ke skupině složené z datlů (Picidae), medozvěstek (Indicatoridae), tukanů (Ramphastidae) a "vousákovitých" ("Capitonidae" – umělá skupina parafyletická vůči tukanům). Výslednou skupinu Fürbringer dokonce pojmenoval, a to jako Pico-Passeres. S Fürbringerem souhlasil i Lowe (1946), který dokonce navrhl včlenit čtyři výše uvedené skupiny do pěvců samotných jako "podřád" Pici. Stejnou fylogenetickou hypotézu podpořili i Mayr & Amadon (1951). Nejobvyklejší alternativou byli "srostloprstí" ("Coraciiformes"). Se vzájemným vztahem šplhavců (Piciformes: Pici + Galbulidae + Bucconidae) a "srostloprstých" to bylo vždycky poněkud komplikované: celá řada autorů pokládala leskovce (Galbulidae + Bucconidae) za blíže příbuzné "srostloprstým" (např. Olson 1985), a naopak dudkovité (Lowe 1946) či klad tvořený dudkovitými, dudkovcovitými a zoborožci (Olson 1985) za blízké příbuzné Pici. Dnes se zdá, že "srostloprstí" jsou skutečně parafyletičtí vůči monofyletickým šplhavcům (avšak opačně, než si myslel Olson; zoborožci a dudci k nim naopak mají dál než zbytek skupiny – viz Hackett et al. 2008), a že tedy dohady, zda mají k pěvcům blíž jedni nebo druzí, nemají žádnou cenu. Nicméně hypotézu o tom, že sesterským taxonem k pěvcům jsou pikokoraciani (zoborožci + datli a všechno mezi tím) jako celek, můžeme považovat za dobrý začátek.
Na konci 60. let a v letech 70. došlo v ptačí systematice ke dvěma významným změnám: nástupu explicitně fylogenetických metod a přesunu od morfologických znaků k molekulárním datům. Tyto dvě novinky se ale dlouho vyvíjely mimoběžně, takže využití parsimonie bylo omezeno na morfologii a molekulární data byla až do 90. let analyzována často sice numerickými, ale relativně primitivními, na distancích založenými metodami typu UPGMA. Snahy analyzovat genetická data distančními metodami vyvrcholily slavnou prací Sibleyho a Ahlquista (1990), která nabízí další hypotézu k otestování. Výše uvedený obrázek ji zachycuje dosti nepřesně, protože zobrazuje jen ty taxony, jejichž pozici testovali také Wang et al. (2011). Sibleyho a Ahlquistovu bichli sice nemám, ale stromek z ní jsem viděl přetištěný v několika jiných publikacích (Cracraft et al. 2004; Livezey & Zusi 2007) a na něm je sesterskou skupinou pěvců klad složený ze většiny tradičních "krátkokřídlých", dále z holubů, dlouhokřídlých, stepokurů, denních dravců ("Falconiformes"), potápek, faetonů, různě rozptýlených "brodivých" a "veslonohých", trubkonosých, tučňáků a potáplic.
Hypotézy, které vyvstaly, když se ke konci 90. let situace přece jen změnila k lepšímu a i molekulární data se začala analyzovat fylogeneticky, jsem už popsal v minulém článku. Zopakuji tedy jen, že dva největší takové rozbory (Ericson et al. 2006; Hackett et al. 2008) shodně odhalily jako nejbližší žijící pěvců papoušky a jako druhé nejbližší příbuzné sokolovité.
Výčet zbývá uzavřít s asi posledním z velkých zdrojů dat, totiž s mitochondriální DNA. Ta v 90. letech podporovala dosti neuvěřitelnou hypotézu, podle níž byli právě pěvci – a nikoli paleognáti – sesterskou skupinou ke všem zbývajícím ptákům (Mindell et al. 1997; Härlid & Arnason 1999), což se ukázalo být jen artefaktem přitahování dlouhých větví (viz níže). Od té doby přišla řada důvěryhodnějších mtDNA analýz. Brown et al. (2008) použili neúplné mitochondriální sekvence (4594 párů bází) od úctyhodného množství taxonů (135) a v jejich výsledcích byli pěvci stále pěkně bazální: vyšli jako sesterská skupina všech ostatních neoavianů; bazálnější tedy byla už jen drůbež a paleognáti. To Pratt et al. (2009) použili kompletní mitochondriální genomy (přes 13 kilobází), ale od omezeného počtu taxonů (45). Vyšel jim pro změnu sesterský vztah s kukačkami, který už dříve analýzou mtDNA odkryli Sorenson et al. (2003) a kombinovaným rozborem morfologických znaků a tří jaderných genů Mayr et al. (2003).
4. Psittacopasserae znovu na scéně
A výsledky? Nic překvapivého. Byly potvrzeny výsledky Hackett et al. (2008), a to nejen v oblasti blízkých pěvčích příbuzných, ale i v topologii "pozemních ptáků" (landbirds – slib napsat o nich něco víc stále trvá...) obecně; jen pozici denních dravců (Accipitriformes) a sov (Strigiformes) nešlo ověřit. Tyto výsledky silně odporují Sibleymu a Ahlquistovi (1990) i topologiím založeným na mtDNA. Livezey & Zusi (2007) se dostali blíž a jejich strom jakousi verzi pozemních ptáků snad i ukazuje; zatímco podle nich ale mezi potomky posledního společného předka sokolovitých a šplhavců patří i Huxleyho cypselomorfové a kukačky, podle Hackett et al. (2008) ani Wang et al. (2011) tomu tak není. Ti druzí dokonce odkryli pozemní ptáky se 100% posteriorní pravděpodobností. Stejná je i pravděpodobnost existence kladu zahrnujícího trogony, zoborožce a dudky (Bucerotes), "jádro srostloprstých" a šplhavce, který rovněž odporuje Livezeyově a Zusiho hypotéze. V ní takový klad zahrnuje ještě pěvce a myšáky.
Wang et al. (2011) dále potvrdili existenci kladu (seriemy + sokolovití + papoušci + pěvci), který spolu s Boydem (2011) nazývám Passerimorphae, a existenci kladu (papoušci + pěvci), který Suh et al. (2011) nedávno nazvali Psittacopasserae. Ten ovšem nemá nijak vysokou podporu: bootstrapovou jen okolo 30% a posteriorní pravděpodobnost (která je obvykle vyšší než bootstrap, takže podporu trochu "nafukuje") 70%. U podobného typu analýz, který využívá zřetězené sekvence mnoha různých lokusů, ovšem můžeme pravděpodobnost nějaké konkrétní topologie určit i jiným způsobem, a to tak, že se podíváme na to, které geny ji odkrývají a které jí naopak odporují. Tento přístup hojně využíval při svém subjektivním hodnocení dosud provedených analýz např. Mayr (2011). Jeho aplikace na nynější studii ukáže, že z 30 lokusů jich celých 28 vykrylo klady Passerimorphae a Psittacopasserae buď v nejvěrohodnějším stromě (= stromě, který je optimální podle metody maximum likelihood), v bootstrapovém konsenzuálním stromě**, nebo v obou z nich.
Rozdíl oproti dřívějším analýzám (Hackett et al. 2008; Suh et al. 2011) spočívá hlavně v tom, že sokolovití zde mají blíž seriemám než papouškům a pěvcům; jinak řečeno, nebyl nalezen klad Eufalconimorphae. V tom se naopak – jak podotkl Alexander Suh na DML – shodují s jednou z dílčích analýz Ericsona et al. (2006), v níž autoři strom zakořenili s drůbeží (Galloanserae) namísto paleognátů. Takový výsledek je sice intuitivní (seriema a sokol jsou si asi podobní více, než sokol a kakadu), ale také krajně nedůvěryhodný. Suh et al. (2011) podpořili Eufalconimorphae 7 retropozonovými inzercemi, největším počtem, jaký v jejich analýze nějaký klad obdržel (jinak řečeno, z hlediska retropozonů máme u monofylii eufalkonimorfů víc důkazů, než např. o monofylii pěvců). Po zahrnutí dat z Hackett et al. (2008) se ovšem Eufalconimorphae znovu objevilo; současně také prudce vzrostla podpora pro Psittacopasserae (na téměř 80%).
Zajímavé také je, že rozdíly v zarovnání sekvencí měly jen velmi malý dopad na výsledky analýzy. Ať už bylo ponecháno původní manuální zarovnání nebo z něj byly odstraněny výše zmíněné problematické úseky, výsledky si stále byly hodně podobné: odstraněné části poskytovaly stejný signál jako ty, které byly ponechány. Stejně tak automaticky vygenerovaná zarovnání stále vyhazovala podobnou topologii, a to s jedinou výjimkou u programu ClustalW2, který je ovšem "known to perform poorly" (Wang et al. 2011:18). Autoři argumentují, že jejich výsledky jednoznačně dokazují, že Chojnowski et al. (2008) měli pravdu, když označili introny za ideální zdroj dat pro fylogenetické analýzy založené na sekvencích, a že výsledky dosud provedených analýz založené na intronech (hlavně pak Hackett et al. 2008) nebyly zkreslené problémy se zarovnáním. Stále ovšem zbývá řada jevů, které mohly výsledky zkreslit, a Wang et al. (2011) se na ně připravili.
Jedním z nich je nechvalně známé přitahování dlouhých větví (Long Branch Attraction, LBA). Taxony, které se z molekulárního hlediska vyvíjejí rychleji než jiné (= v jejichž genomu častěji dochází k bodovým mutacím), budou na dané pozici sdílet tentýž nukleotid s větší pravděpodobností, než taxony s pomalou molekulární evolucí, a to bez ohledu na to, jaké je jich skutečné příbuzenství. Špatnou zprávou je, že zrovna pěvci dlouhou větev skutečně mají, a jak jsem napsal výše, některé fylogenetické rozbory to dokázalo pěkně zmást. Rozbor Wanga et al. (2011) to ale zřejmě příliš neovlivnilo, jelikož pěvci se zde shlukují s taxony, jejichž rychlost molekulární evoluce je pod mediánem: s papoušky, sokolovitými a seriemami.
Jelikož si ale autoři chtějí být jistí opravdu vším, prověřili ještě, zda náhodou přitahování dlouhých větví nemůže aspoň za monofylii Psittacopasserae, protože větev papoušků je pořád delší než větev sokolů a seriem. Metod, jak se s LBA vyrovnat, je několik. Může se použít specifický evoluční model***, který tyto rozdíly bere v úvahu. Wang et al. (2011) jeden takový použili a Passerimorphae i Psittacopasserae byly na výsledném stromě stále přítomny. Pokud je to možné, můžeme taky nahradit taxony s dlouhou větví těmi, jejichž molekulární evoluce je pomalejší. Zde to možné bylo: poměrně "pomalí" jsou bazální pěvec pokřovník (Acanthisitta) a bazální zpěvný lyrochvost (Menura), ale nic naplat – i když odstraníme z matice všechny ostatní, rychlejší pěvce, oba taxony se stále sdružují s papoušky a pak se sokolovitými a seriemami. Že by tato klady byla pouhými artefakty, nenaznačil ani test na saturaci: z té bylo v podezření 8 lokusů, jejichž odebrání opět výsledky nijak neovlivnilo. Závěr je jasný: data podporující blízkost papoušků a pěvců nesou pravý fylogenetický signál.

Bayesiánská analýza 30 manuálně zarovnaných jaderných lokusů, používající model CAT-GTR, který by měl být odolný vůči přitahování dlouhých větví. Na stromu jsou dobře patrné klady z Hackett et al. (2008), včetně Psittacopasserae (papoušci + pěvci), Passerimorphae (papoušci + pěvci + sokolovití + seriemy; zde s poněkud neobvyklou vnitřní strukturou) a Picocoraciae (šplhavci a vůči nim parafyletičtí "srostloprstí"). Čísla nad uzly udávají jejich posteriorní pravděpodobnost. (Zdroj: Wang et al. 2011: Figure 3)
5. Myšáci, biogeografický původ pěvců a implikace pro Metaves
Další zajímavou částí studie je její zjevná neschopnost vypořádat se s myšáky (Coliidae). V článku o retropozonové studii Suha a spol. jsem napsal, že si tento klad "buduje reputaci fylogenetické hádanky srovnatelné i s proslulým hoacinem". Wang et al (2011) tento můj úsudek pouze potvrzují. Vzpomínáte na 2 lokusy, které nepodpořily sesterský vztah mezi pěvci a papoušky? Podle nich byly sesterskou skupinou papoušků myšáci, a teprve potom pěvci. Tento vztah podporuje také morfologie zadních končetin a gen ZENK (Chubb 2004), ostře mu ovšem odporují retropozony (Suh et al. 2011) a dosavadní sekvenční data (Ericson et al. 2006; Hackett et al. 2008). Ostatní geny v datasetu Wanga a spol. myšáky umisťují v sesterském vztahu k trogonům, sokolovitým, pěvcům (14% analýz), nebo psittakopasseranům či passerimorfům jako celku. Po vyloučení myšáků výrazně vzrostla stabilita všech kladů napříč stromem a zvýšila se i bootstrapová podpora passerimorfů a psittakopasseranů. Autoři ale nepovažují situaci z hlediska sekvenčních dat za zcela beznadějnou: problém s myšáky spočívá především v jejich dlouhé větvi, kterou by možná šlo zlomit důkladnějším taxon samplingem: pomoct by mohly třeba sovy (s nimiž se myšáci shlukli v Hackett et al. 2008), které do studie nebyly zahrnuty, jelikož pro příbuzenství pěvců nemají velký význam. Zatím ale nemůže být vyloučena možnost, že myšáci mají blízko k pěvcům – a asi i jakékoli jiné skupině pozemních ptáků.
Wang et al. (2011) se také zabývají biologickými důsledky sesterského vztahu mezi pěvci a papoušky, především vokálním učením. V této pasáži prakticky do detailů opakují to, co už před nimi na toto téma publikovali Suh et al. (2011): dosud se myslelo, že tuto schopnost nezávisle na sobě vyvinuly tři skupiny ptáků (papoušci, zpěvní pěvci a svišťouni), pod novou hypotézou se ale zdá logičtější, že první dvě ji zdědili od společného předka, což podporují i nedávná zjištění vokálního učení u některých křikavých. Zajímavější je to, co autoři o říkají o biogeografii. Sesterskou skupinou vůči zbytku pěvců (zahrnujícímu jak křikavé [Suboscines], tak zpěvné [Oscines]) jsou pokřovníkovití (Acanthisittidae), drobný, málo prozkoumaný klad špatných letců endemických pro Nový Zéland. Základní dělení papoušků je na klad zahrnující naprostou většinu diverzity celé skupiny, včetně kakaduovitých a arů, a na skupinu Strigopidae (česky zřejmě kakapovití), zahrnující největšího žijícího papouška – nelétavého, silně ohroženého kakapo sovího (Strigops) – a jednoho z nejinteligentnějších známých ptáků s extrémně rozvinutou schopností používat nástroje, nestora kea (Nestor). Zajímavé na nestorovi je ovšem také to, že stejně jako pokřovníkovití pochází z Nového Zélandu: že by právě zde žil společný předek celé skupiny Psittacopasserae? Podle Wanga a spol. je to pravděpodobné, vyloučit ale nelze ani to, že diverzifikace celé skupiny sledovala rozpad Gondwany, jak bylo navrženo např. pro "ratity" (Cracraft 2001; Johnston 2011). Molekulární hodiny se ale takovému scénáři zdají odporovat.
Poslední příležitost dozvědět se něco víc o příbuzenstvích pozemních ptáků představuje zkombinování původního datasetu se sekvencemi Hackettové a spol. ve stylu "víc genů víc ví". Hlavním cílem autorů bylo posoudit pomocí analýzy "independent evidence" dosavadní hypotézy; není ale bez zajímavosti z toho vzešlý strom srovnat s "total-evidence" stromem, který už nic netestuje, ale má k dispozici ještě nějaká data navíc. Výsledek ale není příliš překvapivý: topologie se přesně shoduje s tím, co odkryli Hackett et al. (2008) – s jedinou výjimkou, a tou je pozice lelků (Strisores). V kladogramu Hackettové a kolektivu jsou tito součástí Metaves, kontroverzního kladu, který navzdory tomu, že je založen doslova na pár desítkách nukleotidů v jediném intronu jediného jaderného genu, představuje zřejmě nejlepší hypotézu o bazálních divergencích Neoaves, jakou máme k dispozici. Odkryly ji totiž dvě dosud nejobsáhlejší molekulární analýzy (Ericson et al. 2006; Hackett et al. 2008) a žádná ji explicitně nevyvrátila, včetně té retropozonové (Suh et al. 2011). Jenže zatímco podle ní patří lelkové do Metaves a jeřábi, chřástalové či kukačky naopak spolu s pozemními ptáky do Coronaves, Wang a spol. odkryli klad složený z pozemních ptáků a lelků, který kukačky ani monofyletické jádro "krátkokřídlých" nezahrnuje.
Co tohle všechno znamená? Dost možná vůbec nic, jelikož analýze o podobné otázky vůbec nejde a spousta relevantních taxonů chybí, protože nám neříkají nic zajímavého o pozici pěvců (potáplice, dlouhokřídlí, dropové, holubi, stepokurové, svišťouni, mirandorniti a konec konců i spousta pozemních ptáků). Na druhou stranu ale zatím zdaleka nejobsáhlejší molekulární datová matrice vyprodukovala kladogram, který s hypotézou Metaves prostě nelze sloučit. Co se zbytku analýzy týče, klady Passerimorphae a (Picocoraciae + trogoni) mají ještě vyšší podporu než v Hackett et al. (83% a 79% vs. 64% a 71%), zatímco podpora Psittacopasserae zůstává stejná (78% vs. 77%). Důvodem jsou zřejmě opět zlotřilí myšáci. Ani kladogram vzešlý z kombinovaného datasetu nejeví známky přitahování dlouhých větví.

Věrohodnostní (maximum likelihood) analýza datasetu sestávajícího z 49 lokusů, vzniklého sloučením dat z Wang et al. (2011) a Hackett et al. (2008), s myšáky (a) bez nich (b). Čísla nad uzly udávají bootstrapovou podporu; patrné je, že na stromku zahrnujícím myšáky jsou u významných kladů (Psittacopasserae, Passerimorphae, Picocoraciae + trogoni) podstatně menší. Hvězdička označuje 100% podporu. (Zdroj: Wang et al. 2011: Figure 4)
Celkově lze říct, že Wang et al. (2011) ukázali 1), které klady podpořené Hackettovou a kolektivem jsou nejpevnější a které si zaslouží další výzkum (byť bych si dovolil dodat, že vztahy mezi psittakopasserany, sokolovitými a seriemami asi nebudou tak problematické, jak autoři naznačují; 7 retropozonů je prostě hodně); 2) že studie a publikace, které zacházejí se stromkem z Hackett et al. pomalu jako se zjevenou pravdou (Hieronymus & Witmer 2010; Braun et al. 2011; Dyke & Kaiser 2011), což je přístup, který ornitologie zažila naposledy s tapisérií Sibleyho a Ahlquista, vzhledem k současnému stavu znalostí dobře dělají; 3) že Neoaves skutečně nejsou tvrdou polytomií, ale že i tuto mimořádně rychlou radiaci se povede s dostatečným množstvím molekulárních dat rozřešit. Takže nakonec docela optimistické zprávy.
6. Acknowledgments
Za zaslání fulltextu studie děkuji Alexanderu Suhovi a Janu Zrzavému.
*Každý kodon se skládá ze tří párů bází. V tom 3. dochází téměř vždy k substituci – změně "písmena" – častěji než v 1. a v 1. častěji než v 2. Pokud analýza tyto rozdíly nezapočítá, může podávat zkreslené výsledky: třetí kodon je např. vhodný k rekonstrukci mělkých vztahů (tedy těch, kde je společný předek analyzovaných taxonů poměrně nedávný), které nezasahují hluboko do minulosti; ale může podávat zavádějící výsledky u vztahů hlubokých.
**Neparametrický bootstrap je statistický test robustnosti výsledků analýzy. Jeho fylogenetická implementace v zásadě tahá z původního datasetu s opakováním, dokud nevytvoří nový dataset stejné velikosti ("pseudoreplikát"), v němž některé z původních znaků chybějí a jiné jsou zahrnuty vícekrát. Pro každý z množství pseudoreplikátů potom odhadne nový strom. Můžeme se buď dívat na to, v kolika takových stromech se vyskytuje uzel, jehož podporu testujeme, nebo z těchto stromků můžeme sestrojit strom konsenzuální, který může suplovat strom vytvořený na základě kompletní matrice. Velmi hezky popsáno zde.
***Evoluční model je (v tomto smyslu) popisem procesu nukleotidových substitucí – procesu nahrazování různých bází v DNA různými bázemi jinými. Každý aspekt substitučního procesu (např. rovnovážná četnost určité báze nebo relativní rychlost přechodu mezi danými dvěma bázemi) je v modelu reprezentován parametrem, jehož hodnotu analýza odhaduje spolu s tvarem stromu (topologií) a délkami větví (které udávají střední hodnotu počtu změn, k nimž na té které větvi došlo).
Zdroje:
- http://dml.cmnh.org/2011Sep/msg00378.html
- Bonilla AJ, Braun EL, Kimball RT 2010 Comparative molecular evolution and phylogenetic utility of 3’-UTRs and introns in Galliformes. Mol Phylogenet Evol 56(2): 536–42
- Boyd JH III 2011 Taxonomy in Flux Checklist 2.52: Tree View. Accessed September 13, 2011 at http://jboyd.net/Taxo/tree_aves.html
- Braun EL, Kimball RT, Han K-L, Iuhasz-Velez NR, Bonilla AJ, Chojnowski JL, Smith JV, Bowie RCK, Braun MJ, Hackett SJ, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Reddy S, Sheldon FH, Witt CC, Yuri T 2011 Homoplastic microinversions and the avian tree of life. BMC Evol Biol 11: 141
- Brown JW, Rest JS, García-Moreno J, Sorenson MD, Mindell DP 2008 Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biol 6: 6
- Chojnowski JL, Kimball RT, Braun EL 2008 Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene 410: 89–96
- Chubb AL 2004 New nuclear evidence for the oldest divergence among neognath birds: the phylogenetic utility of ZENK (i). Mol Phylog Evol 30(1): 140–51
- Cracraft J 2001 Avian evolution, Gondwana biogeography, and the Cretaceous-Tertiary mass extinction event. Proc R Soc B 268(1466): 459–69
- Cracraft J, Barker FK, Braun M, Harshman J, Dyke GJ, Feinstein J, Stanley S, Cibois A, Schikler P, Beresford P, García-Moreno J, Sorenson MD, Yuri T, Mindell DP 2004 Phylogenetic relationships among modern birds (Neornithes): toward an avian tree of life. 468–89 in Cracraft J, Donoghue M, eds. Assembling the Tree of Life. New York: Oxford Univ Press
- Dyke G, Kaiser G, eds, 2011 Living Dinosaurs: The Evolutionary History of Modern Birds. Oxford: Wiley-Blackwell
- Ericson PGP, Anderson CL, Britton T, Elżanowski A, Johansson US, Källersjö M, Ohlson JI, Parsons TJ, Zuccon D, Mayr G 2006 Diversification of Neoaves: Integration of molecular sequence data and fossils. Biol Lett 2(4): 543–7
- Fürbringer M 1888 Untersuchungen zur Morphologie und Systematik der Vögel, zugleich ein Beitrag zur Anatomie der Stütz- und Bewegungsorgane, Volumes 1–2. Amsterdam: Von Holkema. 1751 p
- Hackett SJ, Kimball RT, Reddy S, Bowie RC, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han K, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Sheldon FH, Steadman DW, Witt CC, Yuri T 2008 A phylogenomic study of birds reveals their evolutionary history. Science 320(5884): 1763–8
- Härlid A, Arnason U 1999 Analyses of mitochondrial DNA nest ratite birds within the Neognathae: supporting a neotenous origin of ratite morphological characters. Proc R Soc B 266(1416): 305–9
- Hieronymus TL, Witmer LM 2010 Homology and evolution of avian compound rhamphothecae. Auk 127(3): 590–604
- Huxley TH 1867 On the classification of birds; and on the taxonomic value of the modifications of certain of the cranial bones observable in that class. Proc Zool Soc Lond 1867: 415–72
- Johnston P 2011 New morphological evidence supports congruent phylogenies and Gondwana vicariance for palaeognathous birds. Zool J Linn Soc doi:10.1111/j.1096-3642.2011.00730.x
- Livezey BC, Zusi RL 2007 Higher-order phylogeny of modern birds (Theropoda, Aves: Neornithes) based on comparative anatomy: II. – Analysis and discussion. Zool J Linn Soc 149(1): 1–95
- Lowe PR 1946 On the systematic position of the woodpeckers (Pici), honey-guides (Indicator), hoopoes and others. Ibis 88: 103–26
- Mayr E, Amadon D 1951 A classification of recent birds. Am Mus Novit 1496: 1–42
- Mayr G 2011 Metaves, Mirandornithes, Strisores and other novelties – a critical review of the higher-level phylogeny of neornithine birds. J Zool Syst Evol Res 49(1): 58–76
- Mayr G, Manegold A, Johansson US 2003 Monophyletic groups within ‘higher land birds’ – comparison of morphological and molecular data. J Zool Syst Evol Res 41: 233–48
- Mindell DP, Sorenson MD, Huddleston CJ, Miranda HC Jr, Knight A, Sawchuk SJ, Yuri T 1997 Phylogenetic relationships among and within select avian orders based on mitochondrial DNA. 213–47 in Mindell DP, ed. Avian molecular evolution and systematics. San Diego: Acad Press
- Olson SL 1985 The fossil record of birds. 79–238 in Farner DS, King JR, Parkes KC, eds, Avian biology, Vol. 8. New York: Acad Press
- Poe S, Chubb AL 2004 Birds in a bush: five genes indicate explosive evolution of avian orders. Evolution 58(2): 404–15
- Pratt RC, Gibb GC, Morgan-Richards M, Phillips MJ, Hendy MD, Penny D 2009 Toward resolving deep Neoaves phylogeny: data, signal enhancement, and priors. Mol Biol Evol 26: 313–26
- Sibley CG, Ahlquist JE 1990 Phylogeny and classification of birds. New Haven, CT: Yale Univ Press
- Sorenson MD, Oneal E, García-Moreno J, Mindell DP 2003 More taxa, more characters: the hoatzin problem is still unresolved. Mol Biol Evol 20: 1484–98
- Suh A, Paus M, Kiefmann M, Churakov G, Franke FA, Brosius J, Kriegs JO, Schmitz J 2011 Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nature Comms doi:10.1038/ncomms1448
- Wang N, Braun EL, Kimball RT 2011 Testing hypotheses about the sister group of the Passeriformes using an independent 30 locus dataset. Mol Biol Evol doi: 10.1093/molbev/msr230











0 komentářů:
Okomentovat
Povolené tagy:
- <b>tučně</b> = tučně
- <i>kurzíva</i> = kurzíva
- <a href="http://pan-aves.blogspot.com/">pan-Aves</a> = pan-Aves
Podporuje $\mathrm{\LaTeX}$ pro matematické vzorce.